The brain does not have a single mode. The same cells, the same synapses, the same brain areas can compute one thing or another depending on the chemical state they happen to be in. Much of my scientific life so far has been an attempt to understand how neuromodulation (by dopamine, in particular) reconfigures neural circuits to enable learning and memory.
The sections on this page are laid out in chronological order. You can also skip to my PhD project.
my Cambridge years
I joined Magdalene College, Cambridge University in 2017 as an undergraduate student, initially studying Psychological and Behavioural Sciences. Taking advantage of Cambridge’s flexible lecture system, though, I constantly sat in lecture halls of the neuroscience, physiology and biology buildings at Downing Site, and within the first year developed an urge to be closer to the es, synapses, and neurochemicals. Drawn to these intricate machineries underlying animal behaviour, I transferred to Natural Sciences (Biological) in my second year.
I could not have asked for a better place to go to university than Cambridge. There I met most of my favourite teachers and mentors. Dr. Paul Brakefield, who supervised my Part IA Evolution and Behaviour paper, told me some of my favourite stories about butterfly eyespot research, and taught me how to sit with a problem, deconstruct and work through it slowly like an evolutionary biologist. And some of my favourite lectures came from Dr. David Parker, who could go on and on about synaptic plasticity in motor circuits in a precise and passionate way, always reaching for the deeper principle. My college tutor Dr. Stuart Martin allowed me complete freedom to follow whatever I found interesting, including philosophy, logic and maths, which became my lasting passions and changed how I think about science.
from genes, proteins to diseases
My first experience in a real laboratory was at the UCL Institute of Neurology at Queen Square, in Dr. Elizabeth Fisher’s group. I was a summer RA funded by the Genetics Society, working on how a protein called ‘cathepsin B’ interacts with amyloid depositions in mice with Down Syndrome-Alzheimer’s Disease: in Down Syndrome, chromosome 21, where the gene encoding the amyloid protein lies, is triplicated, likely contributing to the increased incidence rate of Alzheimer’s in people with Down Syndrome. Dr. Fisher’s idea at the time was that cystatin B (CSTB), also encoded on chromosome 21 and therefore overexpressed in trisomy, might suppress cathepsin B activity and reduce amyloid clearance, which would be a novel mechanism linking Down Syndrome to Alzheimer’s. I spent the summer learning western blots, protein activity assays, plaque counting. Paige Mumford, the Masters student at the time, took me under her wing; Claudia Manzoni and Dr. Karen Cleverley helped me find my footing; Dr. Frances Wiseman taught me how to think about experimental design. Dr. Fisher herself showed me, still a student mostly in psychology at that time, how to think carefully about disease and genetics. We found that while trisomic CSTB levels were indeed elevated in the cortex, this alone was not sufficient to modulate cathepsin B activity, suggesting additional disease-related factors are needed. That work eventually became part of a published paper on Acta Neuropathol. Commun., and I presented my cathepsin B results at the Genetics Society Summer School in Edinburgh that August.

I did not stay in disease research, however. The summer had confirmed that I wanted a life in science, but it also pointed me toward a different scale: not proteins and plaques, but cells and circuits. A paper I had encountered in a Cambridge lecture just before that summer kept pulling me back: Bi and Poo’s 1998 work on spike-timing-dependent plasticity showed that the order and timing of spikes could control enhancement or depression of synapses: temporal order, where the most fundamental and common kind of causality is found, can be encoded as a snapshot by neuronal connections and retrieved to guide future behaviour. This pointed me towards synapses, plasticity, and memory.
‘Patch-clamping is science and art.’

At the end of my second year at Cambridge, I asked Dr. Ole Paulsen for a research position in his lab and was taken in by one of his postdocs, Dr. Tanja Fuchsberger, who taught me two things: how to master whole-cell patch-clamp electrophysiology, and how to think about dopamine and plasticity. At the time, Bittner et al.’s work on behavioural-timescale synaptic plasticity (BTSP) from Jeff Magee’s lab was a major reference point: they had shown that a single dendritic plateau potential could potentiate inputs that were active seconds before or after it, a plasticity window matching the timescale at which behaviour itself unfolds rather than the millisecond precision required by classical spike-timing rules. Tanja was asking whether dopamine might act as a threshold-reducer for this kind of behaviourally relevant potentiation: whether biology uses dopamine (famously connected to pleasure and a heightened state of brain activity) to let even weak associations induce long-term potentiation (LTP). I tested that idea for my undergraduate thesis and found that dopamine did reduce the stimulation intensity required for LTP induction: in the presence of dopamine, inputs too weak to potentiate on their own could now cross the threshold. Dopamine, in other words, was acting as a gain controller for plasticity. My undergraduate thesis was awarded the Bundy Scholarship.

That year crystallised two convictions. Firstly, dopamine can act as a gain controller, a gate, something that changes what circuits can do at any given time. Secondly, I wanted to move from single synapses to populations: to ask not just whether one synapse strengthens, but how a neuromodulatory signal reshapes what an ensemble of neurones computes.
across the pond
I graduated from Cambridge in July 2020 and spent the next year at Scientific American China in Beijing before starting my PhD. I arrived at the Max Planck Florida Institute for Neuroscience in May 2021.

Everything was new. I had come from patch-clamp, which is intimate and slow: once a cell is patched onto, it was one neurone, one pipette, one quiet but intense afternoon. Now at Max Planck, there were surgeries to learn, stereotaxic injections, craniotomies, implantations of various devices into the tiny mouse brain. On top of these, I also started doing optogenetics, multi-unit electrophysiology, two-photon imaging, and various histological experiments including electron microscopy. At the beginning, Raphael Heldman (now Dr. Heldman!) and Kori-Anne Citrin, the senior PhD students in the lab, taught me the procedures, and over time I have had many more mentors to thank throughout the Institute.
The computational side came more naturally, though the learning curve was no less steep. I entered Max Planck a complete novice in coding, having only learned basic Python, MATLAB and R. Through the first year I learned how to do complex data wrangling, visualisation, population analysis, and so on. By year 3, I had already built an end-to-end fullstack pipeline to turn raw, noisy and high-dimensional neural data into interpretable and informative plots. Gradually, the questions that had been forming since the time in Dr. Paulsen’s Lab, about how neuromodulation reshapes what a population of neurones computes, became questions I could actually test.
The first time I saw multiple neurones firing together on a screen, through a Neuronexus silicon probe, marked a dramatic shift in my thinking. In patch-clamp one hears a single cell at a time with extraordinary intimacy. Now there was a population, and with it came a different set of questions and a pull toward computational neuroscience: PCA, LDA, UMAP, the whole vocabulary of dimensionality reduction that lets one ask what an ensemble is doing rather than what a single neurone prefers.
current project
Rapid locus coeruleus dopamine shapes CA1 dynamics for time/distance estimation
This is my dissertation work. I am preparing a manuscript and will defend my thesis in October 2026.
Navigation to learned goals is critical to survival. Once a mouse learns the route from its winter nest to a food post in a snowy field, it repeats that route on a daily basis almost without fail. Remarkably, the mouse can navigate through the route even when a new bout of snow has washed away yesterday’s navigation cues; when environmental landmarks become unreliable, an animal still navigates reliably based on memory alone.
Such navigation has been known to be state-dependent: when an animal is alert and attentive (say, when sensing imminent danger or when in thirst), precise navigation is of greater import than when the animal is relaxed with no immediate goals. This is where my prior experience with dopamine comes in. In recent years, people have confirmed that when animals are alert, a tiny brainstem necleus called the ‘locus coeruleus’ (LC) releases big doses of dopamine into the hippocampus, on which navigation is known to depend. My question, as I arrived at Max Planck Florida Institute, was whether this novel pathway controlled/modulated memory recall precision during navigation.
We simulated the ‘foraging-mouse-in-winter-snow’ scenario mentioned above using a virtual reality set-up. We put an animal on a treadmill, and made it learn the location of a hidden reward (similar to how the food cache location is hidden from the navigating mouse in the snowfield). Each trial lasted around 5–10 seconds, and in order to obtain the reward successfully on a trial, the animal had to rely on its learned trial structure since the VR setting showed only a constant grey corridor without reward cues. In a sense, the mouse’s task was to compare how far (or how long) it had travelled to the trial structure that it recalled, and to decide when to begin licking for reward. With this set-up, I was able to look into the qeustion of whether LC dopaminergic inputs modulated the mouse’s accuracy of navigation.
method summary
The project combines head-fixed virtual-reality animal behaviour, multi-unit electrophysiology, optogenetics and pharmacology for causal manipulation, dual-colour two-photon dopamine, norepinephrine and calcium imaging, Python analysis, and computational modelling. It also has an yet-unpublished branch of machine learning-based axon sorting.
what we found
LC activity marks the start of navigation. A critical component of navigating towards a goal correctly is to note where the journey has started. We found that a major subset of catecholaminergic LC neurones (ones that release dopamine and norepinephrine) burst at trial-start run onset, which marked the start of navigation on each trial. Importantly, the LC response was not explained by running speed or start of generic locomotion.
Phasic LC activity tunes navigation accuracy. Mice exhibit predictive behaviour when navigating learned mazes: they start licking in anticipation of imminent rewards, accompanied by deceleration which aids licking. We therefore used the start of the mouse’s anticipatory licking bout (i.e. their first lick on each trial) as an indicator of where they ‘thought’ the reward was near. Phasic LC activity significantly correlated with first-lick timing on single trials, and more remarkably, by optogenetically amplifying this trial-onset phasic activity, we managed to delay the animal’s first licks without affecting running or reward-consumption: we thus have a signal that causally modulated behaviour on the single-trial level, and that signal is surprisingly coming from a neuromodulatory centre (the LC), which is supposed to act slowly, on much longer timescales. This single-trial specificity is one of the parts I find most striking: tuning a single moment of LC activity was sufficient to shift when the animal began its reward-predictive action.
Phasic LC activity leads to strong, brief and localised dopamine transients in CA1. Local CA1 pharmacology points to dopamine receptors (rather than adrenergic receptors) as necessary for navigational performance. For this reason, I then used two-photon imaging with dLight, a genetically encoded dopamine sensor developed mainly by the Lin Tian group (now also at MPFI), to look at dopamine signals in hippocampal CA1. The results showed that phasic LC activation as well as natural trial-start run onset produced strong, transient and localised dopamine signals in CA1. In addition, around LC axons in CA1, dLight responses are strongest near the axon, fade over micrometre distances, and decay over the next one to two seconds. That is a surprising scale for dopamine in CA1: instead of a millisecond synaptic connection and a slow global bath, it was something in between. The LC-to-hippocampus dopamine pathway is relatively new (Takeuchi et al. [2016] was among the first widely recognised demonstrations) and much of what the field knows about local dopamine dynamics has come from VTA studies. Our study is the first one to show that LC-derived dopamine in the hippocampus can have high temporal and spatial resolutions, connecting it to the rapid (single-trial timescale) behavioural improvement mentioned above.
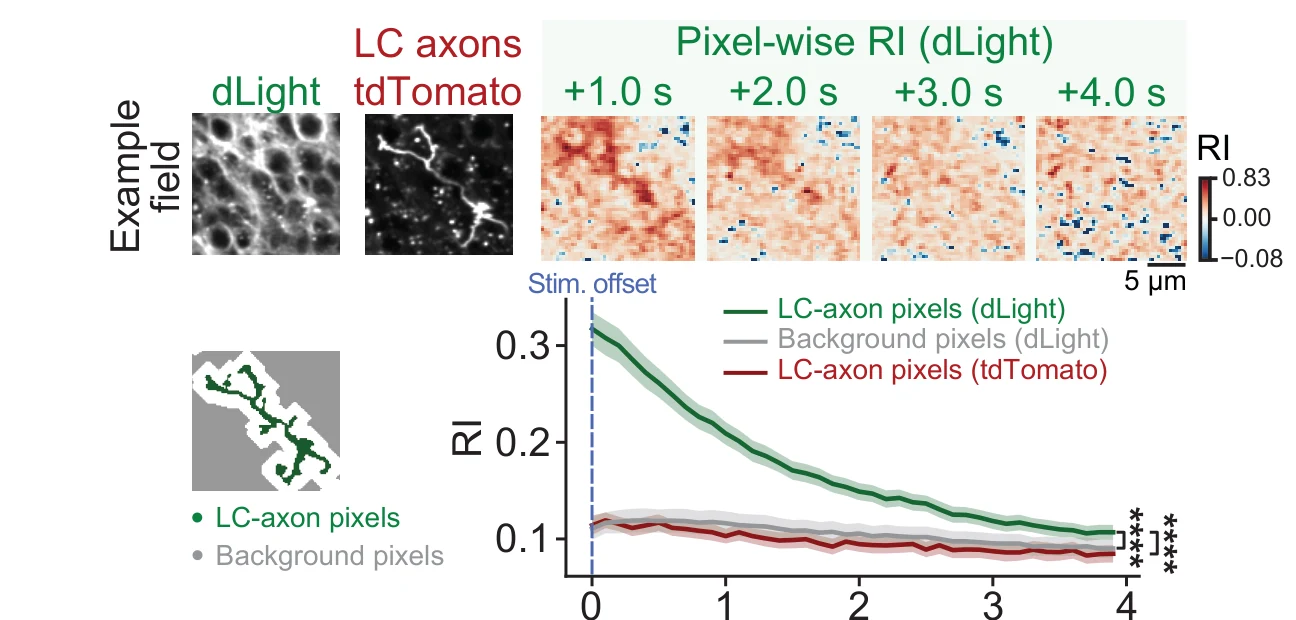
LC-derived CA1 dopamine is linked to a navigation-critical CA1 subpopulation. Dr. Raphael Heldman (an alumnus of the Wang Lab) previously published a paper identifying 2 functional subclasses within the CA1 pyramidal population: PyrUp neurones ramp up at the start of navigation and decay over the following seconds, whereas PyrDown neurones display the opposite activity pattern, rapidly suppressed at navigation onset and ramping back up as the mouse gets closer to the goal. In both Dr. Heldman and my papers, it is shown that PyrUp neurones are important for navigational performance. In particular I have shown that the activity strength of PyrUp neurones during navigation predicted the timing of the mouse’s first lick on the single-trial level. Notably, on the single-trial timescale, phasic LC stimulation increased the recruitment of PyrUp neurones, as well as enhancing their activity on stimulated trials. In addition, by using dual-colour two-photon imaging (thereby monitoring both CA1 neurones’ activity and the dopamine input at the same time), we showed that CA1 neurones with run-onset dopamine signals are likelier to be PyrUp neurones and have stronger ramping dynamics than those without dopamine inputs; blocking dopamine receptors locally, in contrast, reduced the percentage of PyrUp neurones as well as their activity amplitude. The behavioural shift, dopamine signal, and CA1 population change all meet on the same timescale.
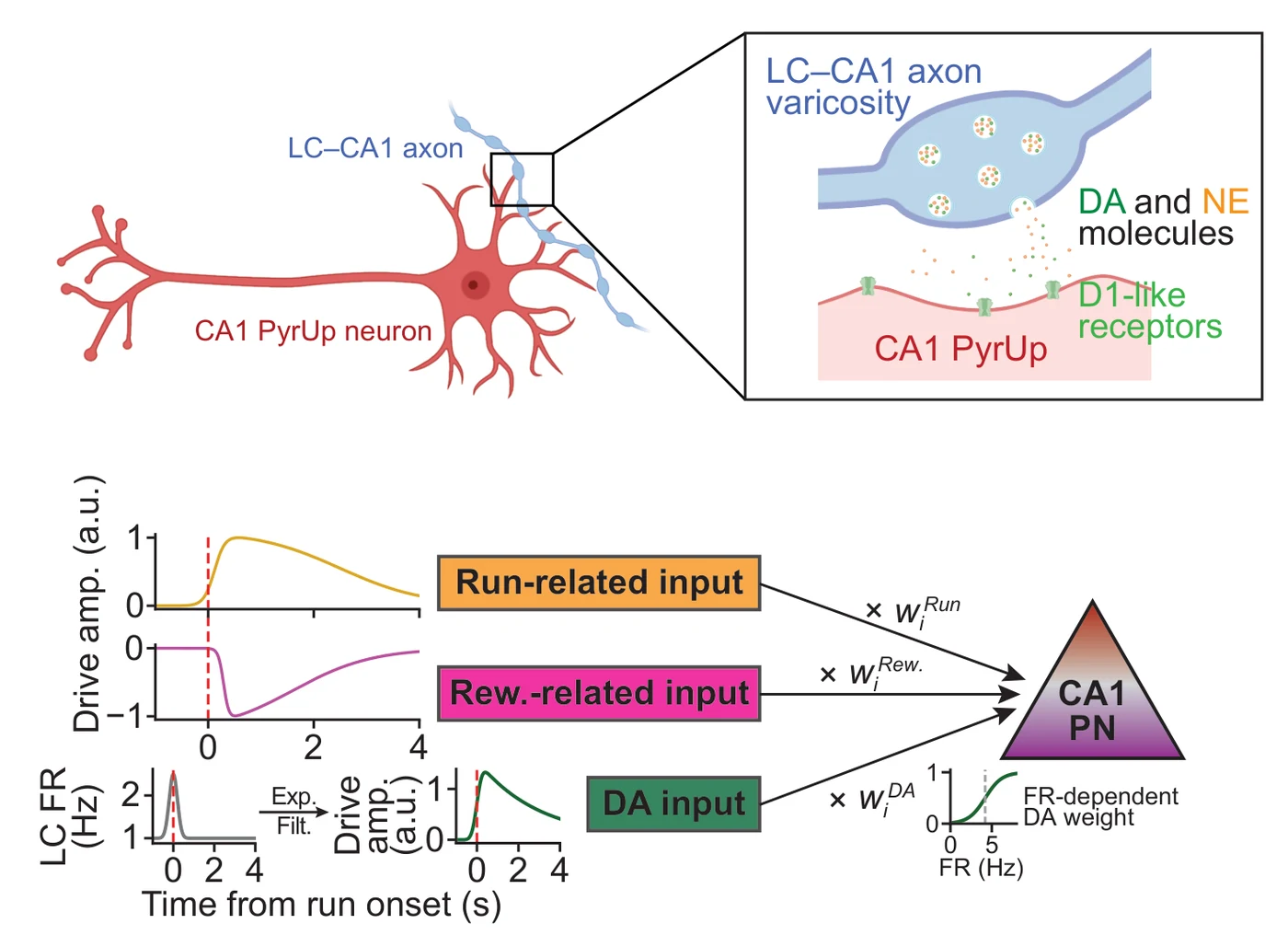
A computational model reproduces experimental observations. After establishing this LC–DA–CA1–behaviour pathway, I built a compact, phenomenological model for the LC–CA1 circuit. The model uses three inputs to a synthetic CA1 pyramidal population: run-related input, reward-related input, and a phasic LC signal transformed into a dopamine drive. The run and reward terms follow Raphael Heldman’s Nature Communications and bioRxiv models of CA1 pyramidal neurone dynamics. The key insight supporting the model was that dopamine did not operate globally on all CA1 pyramidal cells at a prolonged timescale: it acted only upon neurones with high-enough activity at any timepoint, thereby an active neurone that receives dopamine inputs become even more active, whereas an inactive neurone would not be nudged by any incoming dopamine signal. This small addition into the simple model was enough to replicate our main experimental observations.
why this matters
Classical neuromodulation is often treated as diffuse, global, and slow. This has been the idea behind the ‘dopamine = happiness’ notion in popular science for a while: dopamine modulates the state one is in, and provides a general ‘pleasure tone’ but does not associate with moment-to-moment cognitive computations in the brain. My PhD study has shown an intermediate mode: seconds-timescale, micrometre-scale dopamine that prepares/primes a hippocampal computation at the moment a decision begins to unfold. A circuit can ‘shift gear’ quickly and precisely without affecting the tonic state of the whole network.
This also connects back to what I saw in the Paulsen Lab, that dopamine is not just a reward signal but a gain controller. The difference is the scale at which it operates. In the Paulsen lab, the question was whether dopamine lowers the threshold for a single synapse to change. Here the question is whether a phasic dopamine signal, delivered locally and lasting only seconds, can bias what an entire population of neurones does next. The answer, it seems, is yes.
outputs

Manuscript in preparation
Luo D, Cao J, Heldman R, Tian L, Wang Y (2026). ‘A fast, spatially confined dopamine signal from the locus coeruleus tunes hippocampal dynamics to time goal-directed actions.’
Most of the analysis code for my PhD project is available through GitHub.
Selected presentations
- Locus coeruleus dopamine rapidly modulates CA1 dynamics to sharpen goal-directed navigation. COSYNE 2026, selected poster.
- Locus coeruleus dopaminergic modulation of CA1 shapes behavioural timescale dynamics for time integration. Lake Conference, Seattle, 2025.
- An LC-CA1 circuit for initiating path integration. Sunposium, West Palm Beach, 2025.
- Locus coeruleus regulates hippocampus-dependent integration at single-trial level. Memory Formation Forum, Max Planck Florida Institute for Neuroscience, 2024.
publications and code
Wu Y, Mumford P, Noy S, Cleverley K, Mrzyglod A, Luo D et al. (2023). ‘Cathepsin B abundance, activity and microglial localisation in Alzheimer’s disease-Down syndrome and early onset Alzheimer’s disease: the role of elevated cystatin B.’ Acta Neuropathologica Communications 11, 132. doi:10.1186/s40478-023-01632-8
Last updated May 2026